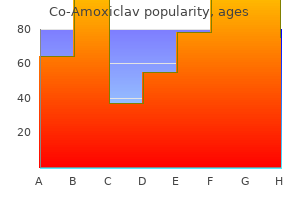
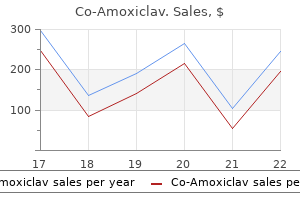
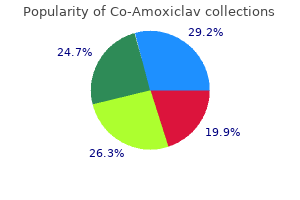
"Buy co-amoxiclav 625 mg with mastercard, medicine for uti".
W. Gelford, M.A., M.D., M.P.H.
Deputy Director, Florida State University College of Medicine
Several strategies are available to detect and to account for the effects of population structure in genome-wide association studies. Quantile-quantile plots of association P values across loci can reveal genome-wide departures that might be attributable to population structure. Population genetic methods can be applied to genotypes at markers throughout the genome to identify groups in the study sample. Investigators can then split the sample into homogeneous groups for association testing or they can include population membership as a cofactor in association tests. Not all population structure is an impediment to detecting genotype-phenotype associations. When populations result from gene flow between genetically divergent lineages, phenotypic differences among these admixed individuals may be attributed to allelic differences Human populations have been the target of most genomewide association studies to date. Second, a subset of these associations replicates in other human populations or involves loci previously known to affect the phenotype (or both). This congruence suggests that some identified associations reflect mechanistic connections between genotype and phenotype. Third, most identified loci were not previously known to affect the phenotype of interest, indicating that genome-wide association testing is a powerful approach for discovering new variants that contribute to evolution. Simulation studies and results from humans suggest that large sample sizes are typically required. Additionally, surveying molecular variation across genomes is a challenging task in organisms without genomic tools. Amassing large numbers of unrelated individuals from a population is an achievable goal, especially for organisms that are easy to collect such as invertebrate animals, plants, and microbes. Although surveying thousands of individuals may not be realistic, sampling hundreds may suffice for finding the loci that contribute disproportionately to trait variation. The genomic resources required for genome-wide association testing can now be developed for a broader variety of organisms. Although this procedure is not cheap, rapid advances in sequencing and genotyping are bringing the cost within reach of evolutionary biologists studying organisms that are not traditional genetic models. Results from the candidate gene approach should provide further motivation for genome-wide association studies in natural populations. This strategy has identified a variety of genes that contribute to variation in evolutionarily interesting phenotypes in natural populations (see chapter V. Although these links have been biased toward genetically simple phenotypes controlled by small numbers of genes, some traits have been more complex, including behavior. A more fundamental challenge is how to proceed after a genotype-phenotype association is identified. Laboratory crosses and association studies do not evaluate the biological mechanism that connects genotype with phenotype. In the future, sequencing genomes and exhaustively testing for associations at all variants (rather than relying on markers) may ameliorate this problem, but the resolution may still be limited by linkage disequilibrium (and the multiple testing burden will only worsen). Creative methods for whittling down the loci exert small phenotypic effects, a finding that has generated considerable attention. Only a small fraction of the genetic variance in the trait that is suspected to exist in the examined populations (estimated from comparisons among relatives) can be explained by summing the effects of detected associations from across the genome. This "missing heritability" problem is probably caused by limited power to detect many mutations with small effects or low frequencies using reasonable sample sizes and common marker alleles. A practical consequence of this challenge is that individual phenotypic values cannot be accurately predicted from genotypes at the loci statistically associated with the trait. An alternative approach to phenotypic prediction that works better in some contexts ("genomic selection") fits the relationship between the trait and all genotyped variants into a single statistical model. For evolutionary studies, the standard genome-wide association strategy is still preferable because it points to specific genes and pathways responsible for phenotypic evolution. Although the vast majority of human studies target disease phenotypes, nondisease traits show similar properties. For example, genetic differences in height are determined by a large number of mutations with small phenotypic effects. Looking beyond humans, published genome-wide association studies are currently biased toward domesticated plants and animals. Recent and intense selection by humans has produced striking phenotypic divergence within or between these species, increasing the power of association testing.
Considering the tree of figure 2, for example, one may assign one rate for all branches to the left of the root, and another for those to Molecular Clock Dating the right. The implementation is very similar to that described for the strict molecular clock discussed above. The only difference is that, under a local-clock model with k rates of evolution, one estimates k 1 extra rate parameters. The local-clock method may be straightforward to use if biological considerations allow us to assign branches to rate classes; however, in general, too much arbitrariness is involved in applying such a model. This approach allows that the rate of substitution may evolve more slowly than the rate of lineage branching, so that closely related lineages will tend to share similar rates. One implementation of this approach, called penalized likelihood, penalizes changes in rate between ancestral and descendant branches while maximizing the probability of the data. A smoothing parameter, l, estimated through a cross-validation procedure, determines the importance of penalizing rate changes relative to the likelihood. Both the likelihood calculation and rate smoothing are achieved through heuristic search procedures. If a probabilistic model of rate change (see below) is instead adopted there is no need for either a rate-smoothing parameter or cross-validation. A model describing substitution rate change over time is used to specify the prior probability on rates, while fossil calibrations are incorporated as minimum and maximum bounds on node ages in the tree. Let x be the sequence data, t the s 1 divergence times (nodal ages) and r the lineage-specific rates. Bayesian inference is based on the posterior probability of r, t, and other parameters (u): f рt; r; ujxЮ / f рx j t; r; uЮf рr j t; uЮf рt j uЮf рuЮ: р1Ю the Bayesian method is currently the only framework that can simultaneously incorporate multilocus sequence information, prior information on substitution rates, prior information on rates of cladogenesis, and so on, as well as fossil calibration uncertainties, to estimate divergence times. In a Bayesian analysis, one assigns prior distributions on evolutionary rates and nodal ages, and the analysis of the sequence data then generates the posterior distribution of rates and ages, on which all inference is based. Here f(t) is the prior probability distribution on times and f(r t, u) is the prior on rates given the divergence times and model parameters, u, while f(x t, r, u) is the likelihood function of the sequence data, x. It should be noted that Bayesian estimation of species divergence times differs from a conventional Bayesian estimation problem, in that the errors in the posterior estimates do not approach zero when the amount of sequence data approaches infinity; indeed, theory developed by Yang and Rannala in 2006 specifies the limiting distribution of times and rates when the length of sequence approaches infinity. The theory predicts that the posterior distribution of times and rates condenses to a onedimensional distribution as the amount of sequence data tends to infinity. Essentially there is only one free variable, and each divergence time is completely determined given the value of this variable; the variable encapsulates all the information jointly available from all the fossil calibrations. Any specific divergence time is obtained as a particular transformation of this single free variable, and the divergence time estimates are completely correlated across nodes. By examining the fossil/sequence information plot (figure 3), which is a regression of the width of the credible interval for the divergence time against the posterior mean of the divergence time, one can evaluate how closely the sequence data approach this limit. This can be used to determine whether the remaining uncertainties in the posterior time estimates are due mostly to the lack of precision in fossil calibrations or to the limited amount of sequence data. If the correlation coefficient of the regression is near 1, then little improvement in divergence dates can be gained by sequencing additional genes. This method thus allows a decision to be made as to whether digging for fossils or doing additional sequencing, or both, would be a better investment of effort. The fossil/sequence information plot for a Bayesian analysis of primate divergence times. Two large nuclear loci are analyzed, using two fossil calibrations derived from a Bayesian analysis of primate fossil occurrence data. The posterior means of the ages for the 14 internal nodes are plotted against the 95% posterior credibility intervals. For example, if the true age of a fossil is larger than a hard maximum bound used in an analysis, the molecular data may conflict strongly with the fossil-based prior, resulting in overinflated confidence in the ages of other nodes. Yang and Rannala (2006) subsequently developed more flexible distributions to mathematically describe fossil calibration uncertainties. These distributions use so-called soft bounds and assign low (but nonzero) probabilities over the whole positive half-line (t > 0). The basic model used is a birth-death process, generalized to account for species sampling with fossil calibration information incorporated into the probability distribution by multiplying the probabilities for the branching process conditioned on the calibration ages and the probability distribution on calibration ages based on fossil information alone. A subsequent Bayesian approach to this problem by Ho and colleagues, implemented in the program Beast, did not use the "conditional" birth-death prior described above, instead multiplying unconditional probabilities, which is incorrect according to the rules of the probability calculus. The effects of this error on inferences obtained using the Beast program is difficult to judge, and the results should therefore be interpreted with caution.
The extent of an experimental adaptive radiation is governed by two features of adaptation: functional interference, and degradation through disuse. Functional interference arises when adaptation to one way of life necessarily leads to loss of adaptation to another. In simple growth media, a universal source of interference is the contrast between rapid but wasteful use of resources and slower but more efficient use. Laboratory cultures are often taken over at first by types 236 Natural Selection and Adaptation this article outlines only a few of the simplest issues tackled by experimental evolution without referring to more complex themes such as metabolic pathways, social behavior, multispecies communities, host-pathogen dynamics, sexual selection, speciation, and multicellular organisms. With the aid of the appropriate model system, there are few fundamental questions in evolutionary biology that cannot be investigated by selection experiments. It may be objected that studying highly simplified laboratory systems may not be relevant to the behavior of populations embedded in highly diverse communities of interacting organisms living in a complex and shifting environment. It is certainly true that particular adaptations, at all scales, from photosynthesis to webbed feet, will require particular explanations, which in many cases cannot be tested experimentally. The justification for experimental evolution, however, is that a distinctive category of evolutionary mechanisms is operating on all characters in all organisms-indeed, in all self-replicating entities-and that it can be elucidated by experiment in much the same way the very different mechanisms governing physiological or developmental processes are being elucidated. We have now acquired a useful understanding of the way these mechanisms operate and the outcomes they produce in simple laboratory systems. The study of more complex systems is only just beginning, and field experiments have seldom even been attempted. Nevertheless, it is clear that the future of the experimental research program lies in greater realism, so that a steadily broader range of evolutionary phenomena will become explicable in terms of clear, testable mechanisms. The Annotated Origin: A Facsimile of the First Edition of On the Origin of Species, ed. They do so at the expense of discarding incompletely metabolized molecules that can be used as substrates by more frugal types as these substances accumulate in the medium. This "cross-feeding" arising from the biological modification of an initially simple medium often leads to the evolution of complex bacterial communities in laboratory microcosms. It is ultimately based on the irreconcilable demands of different metabolic processes, especially the conflict between fermentation and respiration as energy-producing pathways. A specialized type restricted to a particular way of life derives no benefit from being adapted to others. A population that has become adapted to a novel way of life may then be severely impaired if ancestral conditions of growth are restored. For example, green algae possess a carbon-concentrating mechanism that increases the efficiency of photosynthesis by transporting carbon dioxide to the site in the chloroplast where the initial reactions of photosynthesis occur. This is an expensive process that is switched on only when the external concentration of carbon dioxide is low. If lines are grown at elevated concentrations of carbon dioxide, diffusion alone is sufficient to maintain high internal concentrations and the carbon-concentrating mechanism is unnecessary. Consequently, when these lines have been propagated for a few hundred generations, their carbon-concentrating mechanism becomes degraded, and they are unable to function normally when grown at normal atmospheric levels of carbon dioxide. Adaptation to environments that vary in time is quite different: any type unable to survive conditions of growth that recur from time to time will become extinct, and more generally selection will favor generalists able to grow moderately well in all conditions. If specialization is limited by interference between different specialized functions, then generalists will be less well adapted to given conditions than the corresponding specialist, whereas degradation through disuse should be halted by the recurrent change in conditions, and broad generalists with no impairment of function may evolve. When bacterial populations are cultured with fluctuating temperature, or algae are exposed to alternating light and dark, the usual outcome is the evolution of generalists, often with fitness in either environment comparable with that of the corresponding specialists, suggesting that degradation through disuse is a frequent consequence of specialization. Evolution experiments with microorganisms: the dynamics and genetic bases of adaptation. The experimental evolution of specialists, generalists, and the maintenance of diversity. Ingenious partition of processes responsible for genetic change in experimental populations. Written 50 years after On the Origin of Species by the most prominent evolutionary biologist of the day. A geographical distribution in which popu- lations (or species) occur in different locations or habitats (contrast with sympatry). The situation in which selection acts in contrasting directions in two populations.
Common Mononeuropathies Median and ulnar neuropathies are among the most common diagnoses referred to the electrophysiology laboratory. Several techniques have been described that assess slowing of conduction in the median nerve at the wrist. Two techniques are used almost exclusively in clinical practice: the median sensory antidromic technique (stimulating at the wrist and recording from the index finger) and median sensory orthodromic technique (stimulating the median nerve in the palm and recording over the wrist). In the antidromic technique, the recording site is over one of the digits supplied by the median nerve, commonly the second digit (index finger), and the stimulation sites are proximal at the wrist and at the elbow. However, because the antidromic technique involves a longer distance, it is less sensitive to subtle slowing of conduction across the wrist and therefore less sensitive to mild cases of carpal tunnel syndrome. This antidromic technique is usually applied to more severe cases of carpal tunnel syndrome where the median motor responses are already abnormal. In milder cases of carpal tunnel syndrome, the orthostatic or palmar technique is preferred. In the palmar (orthodromic) technique, the stimulation site is the palm and the recording sites are proximal to the wrist and 254 Clinical Neurophysiology at the elbow. This technique is more sensitive for focal slowing because the distal distance is shorter. The sensitivity can be further improved by comparing the median palmar sensory distal latency to the analogous palmar sensory study of the ulnar nerve. If stimulated and recorded over the same distance (typically 8 cm) then the difference in the distal latencies between these two nerves should be 0. Differences in latencies larger than this indicate focal slowing in the median nerve and in the proper setting diagnostic of a median mononeuropathy at the wrist. One additional advantage is that the proximal and distal recordings can be obtained with one set of electrical stimulations, decreasing the number of shocks compared with that of the antidromic technique. However, with the orthodromic technique, the distribution of axons activated at supramaximal stimulation is more variable, resulting in a less reliable amplitude response. In these mild cases of carpal tunnel syndrome the distal latency is more relevant than the amplitude. When a case of carpal tunnel syndrome is suspected, the median motor nerve is often tested first, which helps guide the decision of which median sensory conduction would be most useful to perform. Example of median (upper tracing) and ulnar (lower tracing) palmar sensory studies in mild median mononeuropathy at the wrist. Note that the difference in the peak latencies between the two studies is nearly 1 ms. If there is no slowing of conduction across the wrist in the motor studies then the carpal tunnel syndrome will be mild and the more sensitive palmar orthodromic technique should be used. Various grading scales have been developed to quantify severity of the median mononeuropathy at the wrist using nerve conduction studies. The most common technique for studying the ulnar sensory nerve is the antidromic method, with the recording site over the 5th digit and the stimulation sites proximal at the wrist and above the elbow. Conduction block in the ulnar nerve across the elbow cannot be proven reliably with sensory studies, but if conduction block is present, the proximal sensory amplitudes may be much reduced or even unobtainable. In the case of pure conduction block, the responses elicited with distal stimulation may demonstrate normal amplitudes and latencies. Stimulation above and below the elbow may be helpful in demonstrating focal slowing of conduction across the elbow by isolating the slowing across a much shorter segment. If there is no demyelination and only axonal injury, the only finding on an ulnar antidromic sensory study may be a reduction in amplitude, a finding that is not helpful in localization. If this is the case, additional sensory studies can be used to further localize the lesion. For example, a lesion can be localized to a site above the wrist if the dorsal ulnar cutaneous nerve is abnormal, since it branches from the main ulnar nerve in the forearm. However, the reliability of dorsal ulnar cutaneous responses is less than that of other nerves more commonly tested and this nerve should be compared with that on the contralateral side. If both the ulnar antidromic study and the dorsal ulnar cutaneous sensory nerves are abnormal, the medial antebrachial cutaneous nerve should be tested. Abnormal findings in this nerve suggest a more proximal lesion of the medial cord or lower trunk of the brachial plexus. As with the dorsal ulnar cutaneous nerve, the lateral antebrachial cutaneous nerve should be compared with that of the contralateral side. Peroneal, sciatic, and femoral neuropathies are common diagnoses referred to the electrophysiology laboratory.